Genetic Disorders Among Acadian People
Yves Lacassie, M.D.
Luisa Flórez, M.D.
Introduction
In the early part of their history, the Acadians were a self-contained community with a strong cultural cohesiveness, including a common religion and language. Together with geographical isolation both in Old Acadia (Nova Scotia) and New Acadia (Louisiana), these factors have contributed to an elevated incidence of autosomal recessive disorders. The Acadian population presents a higher than normal frequency of some rare genetic disorders, such as Friedreich ataxia, Charcot-Marie-Tooth disease, Retinitis Pigmentosa, Alström syndrome, Usher syndrome, Tay-Sachs disease, Niemann-Pick disease, and other genetic syndromes.
As clinical geneticists in the Department of Pediatrics, we do not see many of these disorders. Most are evaluated by neurologists, ophthalmologists, audiologists, or other specialists according to their clinical nature. However, we see many patients of Acadian ancestry with other clinical disorders who are referred to our clinics in order to identify the cause (etiology) of the disorder. Identification is crucially important not only for the physician and the family, so that they can receive genetic counseling, but also for the Department of Public Health, so that strategies for prevention of the disorder can be developed.
We recently reviewed our 15-year experience in the Division of Genetics at LSU Health Sciences Center and Children's Hospital regarding the diagnosis and distribution of genetic disorders of Louisiana patients. This review is especially important because Louisiana is one of the 17 states in the country that does not yet have a birth defect registry, and epidemiological studies to determine relationships between cause and geographical distribution of genetic disorders in Louisiana are non-existent. The review we present here is simply a descriptive one, and one that does not attempt to answer epidemiological questions. However, the review gives us the opportunity to answer some questions raised during the public breakout sessions of the Genetics of the Acadian People forums.
Methods
Between November 1986 and December 2000, we evaluated 5,078 patients. Of the 4,569 patients for whom complete demographic information was available, 3,109 were Non-Acadians (68%), 1,272 were Acadians (27.9%), 175 were from other states (3.8%), and 13 were from other countries (0.3%).
To determine the nature of the genetic disorders among these patients, we followed standard methodologies commonly used in clinical genetics. However, we would like to emphasize our use of two unique and original methods: (i) the MultiAxial Diagnostic System (MADS), created by Dr. Lacassie more than a decade ago (Lacassie, 1994; Lacassie 2002) and (ii) the novel etiological classification of diagnoses we recently developed (Lacassie, LaMotta, and Florez, 2001).
MADS. The idea of developing MADS originated from the important need to improve our
ability to compare diagnoses between clinicians and clinical centers, because most
clinical diagnoses are heterogeneous and based on different criteria. MADS defines
four major descriptive categories (axes) that physicians and geneticists can use to
identify a genetic disorder:
- Axis I - phenotypic diagnosis, which basically describes the major findings
- Axis II - pathogenic diagnosis, which tries to identify the underlying mechanism of the disease - in other words, the way in which the primary cause (etiology) leads to development of the disorder (phenotype)
- Axis III - etiological diagnosis, meaning the specific cause of the disorder
- Axis IV - differential diagnosis, comparing and contrasting the disorder with other disorders that closely resemble or share similarities
In our experience, application of MADS has multiple benefits, including:
- more accurate diagnoses
- more homogeneous and reliable diagnostic categories
- better comparisons of diagnoses from different centers, states, regions and countries
- improved understanding of specific clinical definitions
- increased understanding of clinical genetics among health professionals
- increased interest in clinical genetics among health professionals
- improved disorder management and treatment decisions
- enhanced effectiveness of genetic counseling
Etiological Classification. The second unique method we used to study the distribution of genetic disorders in Louisiana involves classification of the diagnoses. We established seven different diagnostic groups as follows:
- Mendelian-Includes those patients with autosomal recessive, autosomal dominant, X-linked dominant, or X-linked recessive disorders, as well as Mendelian disorders not otherwise specified.
- Chromosomal-Includes those patients with chromosomal abnormalities (numerical, structural, complex, and mosaic).
- Other genetic etiologies-Includes patients with contiguous gene syndromes, mitochondrial diseases, triallelic inheritance, epigenetic anomalies, classic sporadic syndromes, syndromes in identification, and a group composed by sequences, associations, polytopic field defects, spectrums, and blastopathies.
- Polygenic/Multifactorial-Includes diseases of complex inheritance caused by multiple genes and environmental factors.
- Unknowns-Includes those patients with only a descriptive (phenotypic) diagnosis and those without a specific diagnosis.
- Non-genetic etiologies-Includes those patients with disorders caused by teratogens, either environmental (such as fetal rubella or fetal alcohol syndromes) or maternal (such as maternal diabetes or lupus erythematosus) acting early during pregnancy and causing malformations in the newborn, as well as post-natal disorders caused by environmental factors (such as meningitis or polio).
- V codes-Includes patients referred for genetic counseling; those in whom no genetic problem is found, being therefore "diagnosed" as normal; and those who present only minor anomalies running in their family or racial group, being consequently considered to have normal familial or racial variation.
We confined our study to patients living in Louisiana. In an effort to determine differences between geographical location and etiology, we divided the state into two sub-populations, the Acadians and the non-Acadians. (See Figure 1 to identify the different parishes composing each division). Acadians comprise 29% (1,272) and non-Acadians compose 71% (3,109) of the patients living in Louisiana. We have patients from all 22 parishes that compose the Acadian region, but the greatest number of patients are residents of the parishes of Calcasieu (19.5%), Lafayette (15%), Terrebonne (10.6%) and Lafourche (7.6%). This bias is explained by the fact that we held periodic satellite clinics in the cities of Lake Charles, Lafayette, and Thibodaux. A considerable number of patients living in Terrebonne Parish attended the Thibodaux clinic due to the proximity. We also held satellite clinics in Monroe, a typically non-Acadian parish, because such distant clinic events are valuable for patients requiring genetic evaluation who live far away from Children's Hospital of New Orleans.
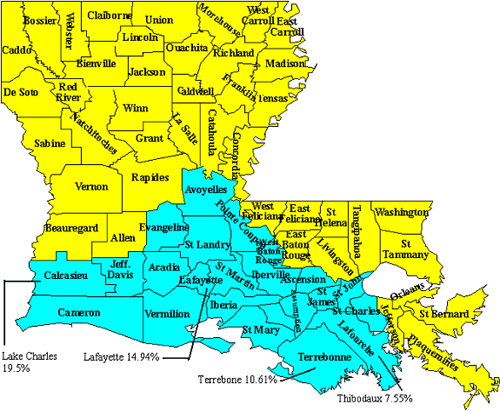
FIGURE 1. A Parish Map of Louisiana, showing the Acadian parishes in light blue. The locations of our satellite clinics are indicated, together with the percentages of patients who reside in the parishes where our clinics were held.
Results
The results of our study provided the answers to common questions about genetic disorders raised during the breakout sessions of the Genetics of the Acadian People forums.
1. Which are the most frequent types of disorders presented? The distribution of etiologies according to our seven-group classification is shown in Figure 2. The largest group is composed by patients with Mendelian disorders (Group 1). The next largest group of disorders (Group 3) is a heterogeneous group including different non-traditional patterns of inheritance and some dysmorphological diagnoses. The third largest group (Group 5) represents a major problem in all genetic centers, as no specific diagnosis is established for particular patients. Polygenic/Multifactorial disorders including common conditions, such as the isolated, non-syndromic cleft lip/cleft palate seen in patients that attended the multi-specialty clinic at Children's Hospital, were seen in 13% (Group 4). Chromosomal disorders (Group 2) were seen in 11%. Only 7% of cases were due to teratogenic (non-genetic) disorders (Group 6), and only 5% of the encounters were for genetic counseling or patients considered to be normal (Group 7). The distribution of these etiologies is similar to distributions reported in studies of other populations.
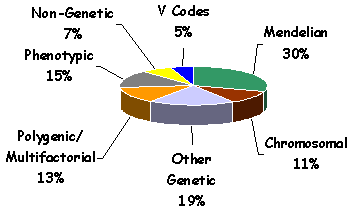
FIGURE 2. The distribution of diagnoses among Louisiana patients according to our seven-category classification method.
2. What are the most common diagnoses that you establish? The most frequent diagnosis is what we call "Syndrome in Identification" (SINID), which refers to those patients who have an evident genetic disorder thought to be due to a single cause. However, at the time of the evaluation, they do not fit into a specific reported, recognizable syndrome. SINID accounts for 13.67% of all patients seen. The second most frequent diagnosis is cleft lip and cleft palate (CL/CP), which accounts for 6.2% of patients seen. Patients with Down syndrome account for 5.22% of the cases. The collection of patients with various metabolic disorders represents 3.9% of the cases. The specific genetic disorder Neurofibromatosis (NF) is the fifth most frequent diagnosis, accounting for 3.7% of the study group. Although we see patients with genetic diseases on a daily basis, we have organized multiple specialty clinics for those most common disorders, such as CL/CP, NF, Down syndrome, and metabolic disorders.
3. Is there any difference in the distribution of diseases between the Acadian and non-Acadian populations? The distribution of disorders among Acadian patients evaluated in our clinics is very similar to the distribution among patients from the rest of Louisiana. The only important differences between Acadian and non-Acadian groups are percentages of (i) non-genetic disorders and (ii) disorders classified as V codes. In the Acadian group, non-genetic disorders account for 4.5% (with teratogenic disorders, mostly Fetal Alcohol syndrome, being 2.2% of the whole group), while 8% of the non-Acadian group are classified with non-genetic disorders (with teratogenic disorders being 4.6% of that group). V Codes accounted for 7% of the Acadians group and only 4.4% of the non-Acadian group. In general, there is a higher percentage of Acadian patients referred for genetic counseling (5.5%) than non-Acadian patients (2.16%).
The distribution of the five most common diagnoses and categories according to our most current review is shown in the following table.
| Diagnosis | Acadian (1,272) |
% | Non-Acadian (3,109) |
% |
| SINID | 134 | 10.53 | 465 | 14.96 |
| CL/CP | 64 | 5.03 | 209 | 6.72 |
| Down Syndrome | 62 | 4.87 | 167 | 5.37 |
| Metabolic disorder | 52 | 4.09 | 121 | 3.89 |
| Neurofibromatosis | 40 | 3.14 | 123 | 3.96 |
4. Have you detected new conditions in the Acadians? Yes. We have diagnosed several new disorders and syndromes in the Acadian population. All new disorders are recorded in our database, and some of these have been presented at meetings or have been reported in medical journals.
We have detected at least three families with different types of microcephaly, two families expressing microcephaly in different autosomal dominant (AD) syndromes and one in an autosomal recessive (AR) syndrome. One of the patients with AD microcephaly (8965) is a girl first seen at 1-1/2 year of age, presenting with a small head, triangular face, pointed chin, thin nose, and prominent and anteverted ears. Her mother and other relatives have similar features. We have named this disorder AD microcephaly Lake Charles type. The patient with AR microcephaly (9433) was seen at 30 months of age. He presents a small head, small ears, anteverted nares, small umbilical hernia, diastasis of the recti (the muscles in the abdomen are separated from each other, leaving a vertical gap between them), and several birthmarks. The parents of patient 9433 are second cousins, so consanguinity is indicated.
Years ago we diagnosed and published the first American patient with Satoyoshi syndrome (Ehlayel and Lacassie, 1995), a resident of Cajun country. Satoyoshi syndrome is a rare disorder of unknown cause that affects multiple systems. Satoyoshi and Yamada first described this syndrome in 1967, and to date most cases of this syndrome have been reported in Japan. The affected patient (3268), who was first seen at age 19 because of short stature, presented fractures and striking bone deformities secondary to muscle spasms. She had lost her hair between age 9 and 10, and she was amenorrheal. Previous evaluations had not established the diagnosis.
Another patient (2259) presents a new, private syndrome. This patient has a complex disorder of unknown etiology. At birth he presented situs inversus abdominalis (the abdominal organs are located in inverted positions, with the organs that must be on the right side located on the left and vice versa), dextrocardia (the heart is located on the right side instead of the left), and umbilical and inguinal hernias. The facial features included blepharophimosis (fused eyelids), micrognathia (small chin), remnant epicanthal folds, and upturned nostrils. He had also wide-set nipples, a brownish spot on the left arm, and bilateral clubfoot. He developed a metabolic disorder with low calcium and seizures (pseudopseudohypoparathyroidism). At age 40 months, he presented decreased muscle tone, mild mental retardation, and brittle enamel of the teeth. He was re-evaluated at age six because he had problems swallowing and breathing, was overweight, and showed insensitivity to pain. On physical examination, he showed undersized genitalia that was deeply sunken in the prepubic fat. There was a familial history of consanguinity: the father of the boy is the mother's uncle, raising the possibility of an autosomal recessive pattern of inheritance.
Conclusion
We present a descriptive review of our 15-year experience (1986-2000) in providing
genetic services to over 5000 patients. Although the distribution of different types
of disorders is very similar in the Acadian and non-Acadian populations, there are
some important differences. One difference is the higher percentage of Acadians than
non-Acadians being referred for genetic counseling; the other is higher frequency
of teratogenic disorders (mainly Fetal Alcohol Syndrome) in the non-Acadian population.
The most common disorders seen in Acadians are similar to those seen in other populations.
Our experience and methodology used have allowed us to establish a very good percentage
of specific diagnoses, to identify new syndromes, and recognize some very rare anomalies,
including the first case of Satoyoshi syndrome published in America.
References
Ehlayel M, Lacassie Y. 1995. Satoyoshi Syndrome: An Unusual Postnatal Multisystemic
Disorder. Am J Med Genet, 57:620-25.
Lacassie Y. 1994. An International MultiAxial Diagnostic System in Clinical Genetics. In Bartsocas, C.S. and Beighton, P. (edit.) In: "Dysmorphology and Genetics of Cardiovascular Disorders." Zerbinis, Athens, (pages 28-31).
Lacassie Y. 1998. Approach to Clinical Diagnosis of Genetic Disorders. BioMedicina 1(2):9-15.
Lacassie Y, Arriaza M. 1998. Clinical Approach to Diagnosing Craniofacial Anomalies. In OMS Knowledge Update: Self-Study Program, vol 2, Craniofacial Section, Dale J. Misiek ed, Am Assoc Oral and Maxillofacial Surgeons, (pages 19-29).
Lacassie Y. 2000. Use of the MultiAxial Diagnostic System in Birth Defects Registries. 3rd Annual Meeting National Birth Defects Prevention Network: Advances and Opportunities for Birth Defects Surveillance, Research, and Prevention, New Orleans, Louisiana.
Lacassie Y, LaMotta I, Florez, L. 2001. Classification and distribution of genetic disorders: The LSU New Orleans/Children's Hospital Experience (1986-2000). Annual Clinical Genetics Meeting (ACMG/32nd Annual March of Dimes Clinical Genetics Conference), Miami, FL (:81).
Lacassie Y, LaMotta I, Florez L. Nosology of genetic disorders. 2001. Eur J Hum Genet (Suppl 1) 9:302(A). 10th International Congress of Human Genetics, Vienna, Austria.
Lacassie Y. 2002. Use of a MultiAxial Diagnostic System in Clinical Genetics (L). Genet Med 4:95-96.
McKusick VA. 1998. Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders. 12th ed. Baltimore: John Hopkins University Press.
McKusick VA. Online Mendelian Inheritance in Man (OMIM). www.ncbi.nlm.nih.gov/omim/
Oxford Medical Databases (London Dysmorphology Database). London: Oxford University Press; 2000.
Rimoin D, Connor M, Pyeritz R. 1996. Principles and Practice of Medical Genetics. 3rd ed. New York: Churchill Livingston.
Scriver C, Beaudet A, Sly W, Valle D. 2001. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill.
About the Authors
Yves Lacassie, M.D., is Professor of Pediatrics and Head of the Division of Clinical
Genetics in the Department of Pediatrics at LSU Health Sciences Center. He is Director
of Genetic Services at the Children's Hospital of New Orleans. A graduate of the University
of Chile, Dr. Lacassie is board-certified in Clinical Genetics. His major interests
are dysmorphology, the delineation of new syndromes, nosology of genetic diseases,
cytogenetics, and the clinical use of dermatoglyphics.
Luisa Florez, M.D. is a Research Fellow in the Division of Genetics, Department of Pediatrics, LSU Health Sciences Center. Dr. Flórez received the M.D. degree at University of Cauca, Popayán, Colombia, and a degree as specialist in Clinical and Anatomic Pathology from University of Valle, Cali, Colombia. After a one-year honorary fellowship in Neuropathology in the Department of Pathology and Laboratory Medicine at Tulane University, Dr. Flórez joined LSU. Her major interest is the area of Neurogenetics.
How to Contact
Yves Lacassie, M.D., FACMG
Professor of Pediatrics
Head Division of Clinical Genetics
Department of Pediatrics
Louisiana State University Health Sciences Center
1542 Tulane Ave., T8-1
New Orleans, LA 70112-2822, USA
Phone: 504-568-6225; FAX: 504-568-7532
e-mail: ylacas@lsuhsc.edu,
or
Yves Lacassie, M.D., FACMG
Director Genetics Services
Children's Hospital New Orleans
Ph: 504-896-9254; FAX: 505-896-3997
e-mail: ylacassi@chnola.org
Dr. Luisa Florez
Department of Pediatrics
Louisiana State University Health Sciences Center
1542 Tulane Ave., T8-1
New Orleans, LA 70112-2822, USA
email: lflore@lsuhsc.edu