|
|
Doug Johnston, PhDAssistant Professor - Research 533 Bolivar Street |
Degrees |
BS, Microbiology MS, Microbiology PhD, Biological Sciences |
Bio |
Dr. Johnston began his research career as a Master's student in the Division of Infectious Diseases at Harbor-UCLA Medical Center in Torrance, CA. Upon graduation, he was invited to continue his work there as a Research Associate, under the tutelage of Dr. Scott Filler. Research in the Filler lab was focused on two medically important fungi, the human pathogens Candida and Aspergillus. Utilizing techniques from biochemistry, molecular biology, and immunology, Dr. Johnston helped to identify and characterize many novel aspects of pathogenesis and drug resistance in these two genera. After several years at Harbor-UCLA, Dr. Johnston switched gears and began his PhD studies in the lab of Dr. Chris Hughes at UC Irvine where the focus was vascular biology, particularly the mechanisms involved in inflammation, wound healing & angiogenesis. Dr. Johnston's research helped to identify numerous components of the signaling pathways that are differentially regulated upon endothelial cell exposure to inflammatory cytokines, including TNF-a, that are important for maintaining vascular integrity and controlling outgrowth. Dr. Johnston joined the Department of Microbiology, Immunology and Parasitology in 2008 when, for his postdoctoral work, he joined the lab of Dr. Joy Sturtevant- coming full circle back to the field of fungal pathogenesis. In 2010, after helping to characterize many aspects of mutant Candida biology, Dr. Johnston accepted an appointment as Assistant Professor-Research where he is now working to characterize the molecular interactions between pathogenic fungi and the endothelium. |
Research Interests |
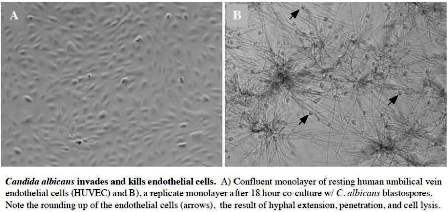
The Johnston lab is focused on defining mechanisms of fungal pathogenesis, particularly the human host response to bloodstream infection by the opportunistic pathogen, Candida albicans. The severity of candidiasis ranges from relatively benign superficial infections to fatal systemic disease. In fact, C. albicans is the most commonly isolated fungal pathogen among severely immunocompromised patients and is the fourth most common cause of all nosocomial infections. Although a common commensal, changes in host immune status allow Candida to penetrate the natural barriers to infection, such as the skin or mucosa, where it gains access to the bloodstream. Once bloodborne, Candida is capable of escaping the vasculature and invading nearly every tissue of the body. Mortality attributed to disseminated candidiasis is often greater than 50%, even with aggressive antifungal treatment. Importantly, for Candida to exit the bloodstream, it requires significant interaction between the fungus and both the endothelial cells lining the vascular wall and the underlying extracellular matrix. We use molecular, genetic, biochemical, immunochemical, and cell-based approaches to define the human endothelial cell response to adherence, invasion, and damage by C. albicans. Our most recent results suggest that invasion by C. albicans results in widespread endothelial apoptosis, loss of vascular barrier function, and the suppression of components of the normal wound healing response, including endothelial migration, proliferation, and differentiation. Vascular dysfunction in this setting likely provides Candida a greater window of opportunity for the establishment of deep tissue infection. We believe that clinical manipulation of the endothelial response to Candida will help to preserve vascular barrier function and lead to enhanced immunity. Our goal therefore is to define the key mechanisms involved in the host endothelial response to Candida and to identify novel potential targets for the development of new antifungal therapies.
|
Selected Publications |