
 Quicklinks▼
Quicklinks▼ myLSUHSC
myLSUHSC
Christopher M. Taylor, PhD
Christopher M. Taylor, PhD
Professor
Department of Microbiology, Immunology & Parasitology
Director
Bioinformatics, Biostatistics, & Computational Biology Core
Louisiana Biomedical Research Network
Professor
Department of Microbiology, Immunology & Parasitology
Director
Bioinformatics, Biostatistics, & Computational Biology Core
Louisiana Biomedical Research Network
533 Bolivar Street, Room 605
New Orleans, LA 70112
E-mail:
§ Curriculum Vitae §
Degrees
Ph.D. in Computer Science
Algorithmic Analysis of Human DNA Replication Timing
University of Virginia, Charlottesville, VA - 2008
M.S. in Computer Science
A Mathematical Model for Knowledge Acquisition
University of Virginia, Charlottesville, VA - 2002
B.S. with Honors in Computer Science and Mathematics
University of Mary Washington, Fredericksburg, VA - 2000
Bio
Dr. Taylor earned his PhD in 2008 from the University of Virginia under the direction of Dr. Anindya Dutta (Harry F. Byrd Professor and Chair of Biochemistry and Molecular Genetics) and Dr. Gabriel Robins (Professor of Computer Science). For his dissertation, Dr. Taylor developed a method for generating a continuous profile of DNA replication timing from discrete pools of replicated DNA hybridized to genome tiling microarrays. This work also proposed a method for finding origins of replication and discovering regions of the genome where alleles replicate asynchronously. This approach was presented at Pattern Recognition in Bioinformatics 2008 in Melbourne, Australia and later published as part of an invited chapter for Methods in Molecular Biology in 2009. During this time, Dr. Taylor was a member of the NIH ENCODE Consortium and a member of the Chromatin, Chromosomes & Replication Analysis group as part of the ENCODE Science publication. He was also the lead analyst for replication and a member of the Integrated Analysis and Manuscript Preparation group for the ENCODE Nature publication.
After his graduate work, Dr. Taylor joined the University of New Orleans as an Assistant Professor of Computer Science in 2008. As a new faculty member, Dr. Taylor shifted his focus from microarrays to the emerging technology of high throughput sequencing. He established several collaborations with basic scientists and clinical researchers in the area and developed two major branches of his subsequent research: Microbial Community Sequencing and RNA Sequencing. This work led to the development of PARSES and RNA CoMPASS, integrated systems for dual RNA-Sequencing analysis along with a software framework for microbial community profiling.
Dr. Taylor joined LSUHSC in December 2012 as an Associate Professor in the Department of Microbiology, Immunology and Parasitology. He built an informatics laboratory at the School of Medicine focused on microbial community sequencing, analysis, and visualization and was appointed in July 2016 as the Director of the Bioinformatics, Biostatistics and Computational Biology Core of the Louisiana Biomedical Research Network. Dr. Taylor was awarded Tenure from LSUHSC in 2017 and was promoted to Professor in 2025.
Clinical Interests
Dr. Taylor's lab has a strong interest in the interaction between reproductive tract microbiota and sexually transmitted infections. He was awarded a Multi-PI R01 from NIH/NIAID in August 2017 to study the consequences of vaginal microbiota on IFNγ mediated clearance of Chlamydia infections. Hi is also closely involved as a co-invesigator with an NIH/NIAID R01 funded in January 2020 to study the relationship of STI screening with adverse birth and newborn outcomes. He is a co-investigator studying the releationship between vaginal microbiota and biofilm formation in bacterial vaginosis on an NIH/NIAID R01 that was funded in February 2020 which is following changes in the vaginal microbiota occuring prior to incident bacterial vaginosis. Dr. Taylor also has clinical interests in the use of shotgun metagenomic sequencing of the reproductive tract microbiota to identify pathogens and direct targetted treatment of STIs.
Research Interests
Dr. Taylor’s research lies in the realm of Computational Biology and Bioinformatics, specifically related to applications of high throughput sequencing. This research is highly collaborative in nature involving the development of algorithms for analysis and visualization of sequencing data. The human microbiome is a specific application of interest and his lab has been involved with studies of the vaginal, gut, oral, and lung microbiomes.
Dr. Taylor's group develops algorithms and tools for analysis and visualization of microbial communities in the context of the human microbiome. This analysis utilizes high-throughput sequencing to assay the composition of the bacteria living within and upon a host organism. Changes in this population have implications in general human health and disease and the analysis of this diverse microbiota presents unique computational challenges.
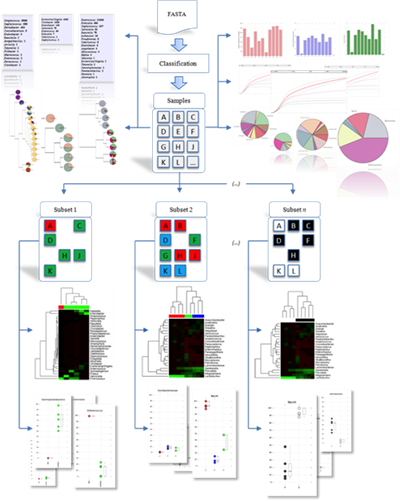
Dr. Taylor's research interests currently focus on understanding of dynamic changes in the vaginal microbiota associated with various STIs including BV, Chlamydia and HIV. He has served as multi-PI on R01 and R21 grants from NIAID, and as a co-investigator on two R01s, an R21, an UL1, and a P20. In addition he served as the contact PI for an NSF grant that was utilized to acquire and establish a high-performance computing cluster called Tigerfish for computational biology and bioinformatics research. Further information on this institutional research resource is available here: https://www.lsuhsc.edu/admin/it/hpc/default.aspx
Software Tool Development
Members of the Taylor Lab develop a variety of software tools to facilitate our research. In the past we have developed systems for microbial community analysis and contributed to development of tools for metagenomic predictions. The latest tool has been developed by John Lammons to faciliate usage of the VALENCIA classifier with Phyloseq in the R environment in order to perform community state type classifications of vaginal samples. More information and download of the tool are available here: https://github.com/JLammons14/ValenciaInR
Teaching Activities
Former Students
Jacob Elnagger - MD/PhD, March 18, 2024: Investigating the temporal dynamics of the vaginal microbiome prior to incident bacterial vaginosis
Clinical Rotations at LSUHSC-NO
Vincent Maffei - MD/PhD, April 2, 2019: Biological Aging: The Role of the Gut Microbiota, HIV, and Alcohol Use Disorder
Gastroenterology Fellow at Wake Forest University
Guorong Xu - PhD, June 18, 2012: RNA CoMPASS: RNA Comprehensive Multi-Processor Analysis System for Sequencing
Principal Bioinformatics Engineer at BioNTech US
A. Murat Eren - PhD, April 6, 2011: Assessing Microbial Diversity Through Nucleotide Variation
Professor of Ecosystem Data Science at the Helmholtz Institute for Functional Marine Biodiversity
Joseph Coco - MS, April 11, 2011: PARSES: A Pipeline for Analysis of RNA-Sequencing Exogenous Sequences
Senior Application Developer at Vanderbilt University Medical Center
Jonathan Brown - BS, May 18, 2012
Senior Software Engineer for Toptal
Eugene Blanchard IV - BS, May 17, 2013
Director of Development for Bus Patrol
Committees & Administrative Responsibilities
Search Committee for Department Head of Genetics, 2013-2014
School of Medicine Research Advisory Committee, 2013-Present
School of Medicine International Travel Committee, 2014-Present
Basic Science Delegate to Faculty Assembly, 2015-Present
Basic Science Delegate for Administrative Coucil, 2016-2017
President of School of Medicine Faculty Assembly, 2018-2019
Representative to Faculty Senate, 2018-2021
Selected Publications
In The News
In January 2015, Dr. Taylor was selected as the winner of $5,000 of Illumina sequencing reagents in Illumina's MiSeq My Focus contest. His lab uses the Illumina MiSeq to explore microbial diversity in the gastrointestinal tract, urogenital tract, airways, and hospital environments. Differences in bacterial community structure in these environments often correlate with diseases and we aim to advance diagnosis and treatment of a variety of conditions including sexually transmitted infections, obesity, diabetes and cancer.